解析基因调控网络GRN中TFs+500数据集
对于GRN任务,作为benchmark的TFs+500/1000数据集是绕不开的。但是,一旦分析一下这个名字,就会发现这个数据集非常诡异。
正如下图所示,你会发现gene列也不是500,也不是TFs+500。

为了解答这个疑惑,不得不去查看其论文。遗憾的是,论文对于这一部分写的非常简略。但是,这篇论文的代码以及数据集都是公开的。并且,我在github issue找到了相关信息:
where is the tfs+500 and tfs+1000 datasets?: https://github.com/Murali-group/Beeline/issues/98
根据回复,我下载并运行了相关代码。
code: https://github.com/Murali-group/Beeline
dataset: https://doi.org/10.5281/zenodo.3378975
整体架构
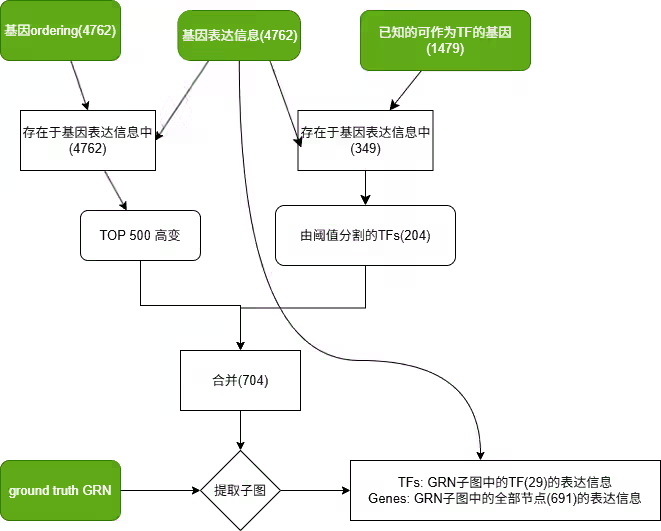
根据代码中的描述,TFs+500 mHSC-E数据集的生成过程如下(括号中的值表示这一步的基因数量):

输入是4个部分:
- gene expression data
- knows TFs
- ground truth GRN
- gene ordering :大致表明了这个基因有用的程度。
你可能发现了,TFs与500个基因的并集刚刚好是500+204=704个基因,没有出现重复的基因。这是因为代码的逻辑是这样的:
1 | variable_genes = [] |
因此,不可能重复。
看了这个流程图,诡异的部分就解开了:输出中的TFs与名字中的TFs+500根本不是一个东西。名字中TFs+500指的是图中的由阈值分割的TFs,以及500个剩下的高变基因。最后输出中的TFs指的是GRN子图中的TFs,而gene指的是全部基因(TFs+500)。
gene ordering
这里的源代码使用R语言实现的,这个我不太熟,让AI帮着看的。
gene ordering的生成过程大致如下
数据预处理
- 对数据进行标准化、对数化
- 过滤表达量太小、残缺的基因
- 扩散映射降维
伪时间值计算
使用Slingshot实现。(括号中是shape)
输入是上述降维后的细胞坐标(n_cells, n_dimensions)、聚类标签(n_cells,)源代码中使用的是细胞所处的真实时间点。
最后得到伪时间值(在指主轨迹分化的程度)(n_cells,),用于后续的分析。(每个细胞对应一个伪时间值)
GAM(广义加性模型)
这个模块计算两者相关的显著性。
输入基因表达信息、伪时间值
输出p值,表示两者相关的显著性。p值越小,两者相关的显著性越高。
排序
p值越小,代表这个基因越有用。p值越大,可能说明这个基因是噪音。
最后将结果升序排列,得到基因的排序。